
XtalOpt is a free and truly open source multi-objective evolutionary algorithm designed for a priori prediction of functional materials with a fixed or variable composition.
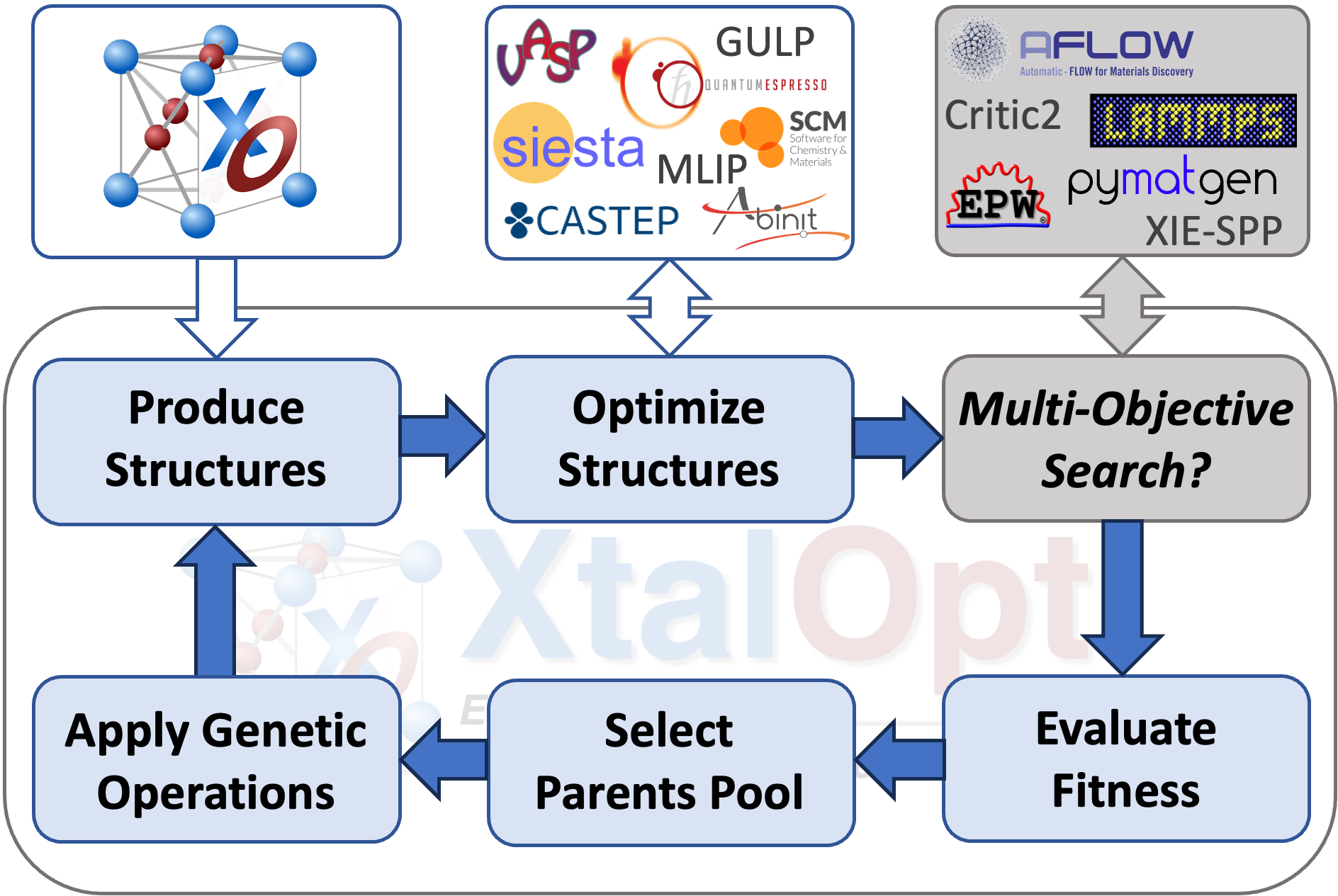
Using objectives (properties) calculated by external codes, XtalOpt can perform multi-objective ground state search using generalized fitness function or Pareto optimization.
About
XtalOpt is an open-source code, published under the "NEW" BSD License, designed to perform multi-objective evolutionary ground state search for novel functional materials over a fixed or variable composition. Local optimizations can be performed using a variety of explicitly supported first-principles codes and classical force fields (e.g. VASP, Quantum-Espresso, Abinit, and GULP); while any desired total energy calculation tool (e.g., machine learning potentials) can be utilized through scripting.
XtalOpt is developed in Eva Zurek's group in University at Buffalo by (in alphabetical order):
- Patrick Avery
- Zackary Falls
- Samad Hajinazar
- Allison Vacanti
With questions, bug reports, and collaboration proposals please contact Eva Zurek at ezurek-at-buffalo.edu.
News
❒ July 18, 2025: XtalOpt Version 14.2 Released
- Option to specify the "minimum number of atoms" in the produced cells, applicable to variable-composition search is added.
- Fixes and improvements are introduced to the workflow of genetic operations.
❒ July 12, 2025: XtalOpt Version 14.1 Released
- The "multi-cut" crossover operation is added, where the parent structures are sliced into a user-specified number of ribbons for a better sampling of offspring atoms in variable-composition search and parents with diverse unit cell sizes.
- The variable-composition search option is possible for elemental systems.
- The experimental option introduced in GUI to Import/Export run parameters from/to XtalOpt CLI input file.
- The crossover for fixed/multi-composition searches improved for better handling of parent structures with large difference in atom numbers.
- A more effective volume adjustment for new structures is applied.
- Relevant to variable-composition search, the user can specify the minimum number of atoms for new unit cells.
❒ June 9, 2025: XtalOpt Version 14.0 Released
- XtalOpt can perform variable-composition search.
- Pareto optimization scheme is available for multi-objective searches.
- Volume limits can be specified for elements.
- Explicit support for moment tensor potentials (MTPs) is added.
- New similarity check option (RDF) is available.
- New genetic operations are added: permutomic, permucomp, and supercell.
- Various bug fixes and improvements.
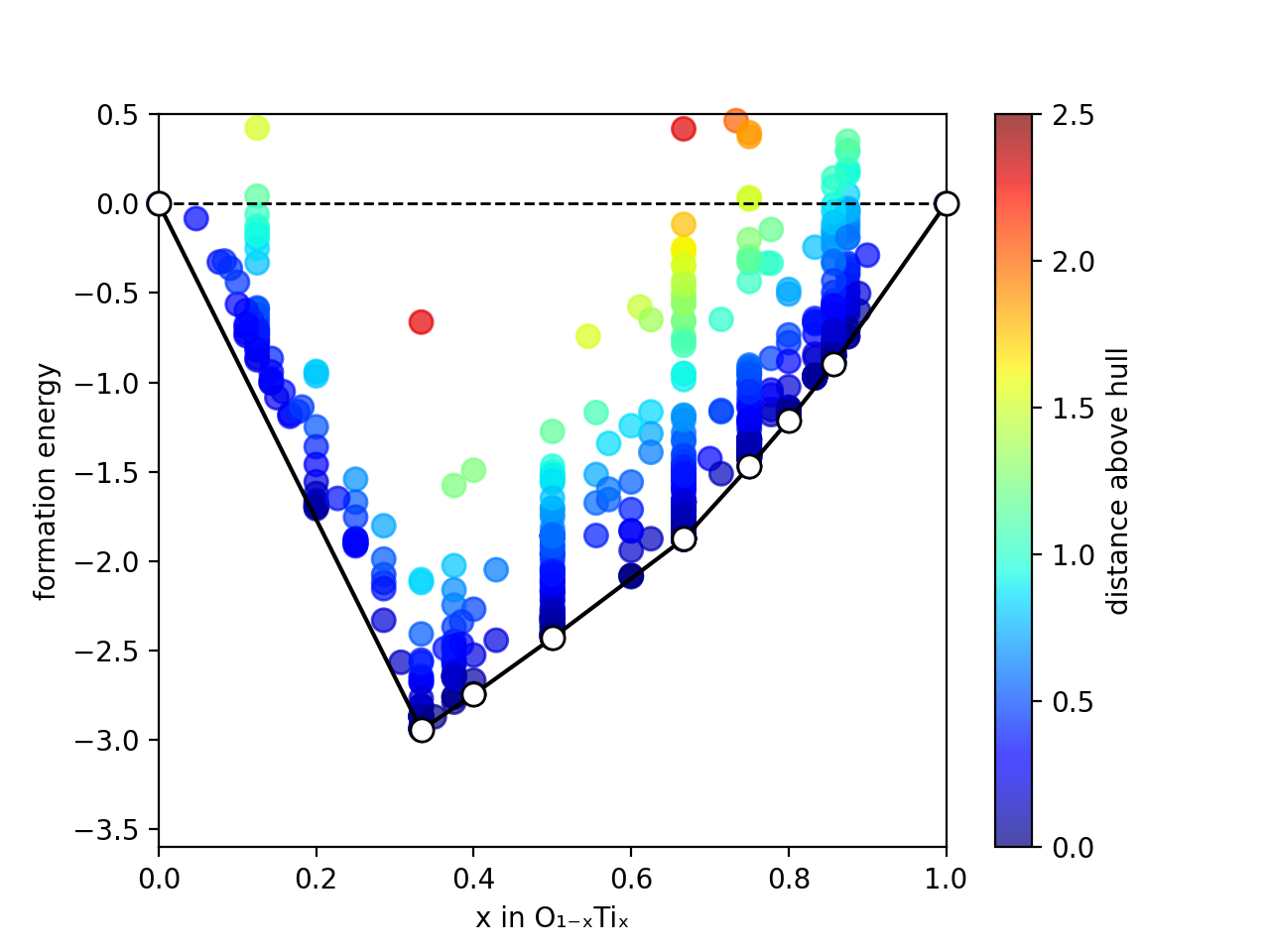
Convex hull of the Ti-O system generated through variable-composition option of XtalOpt 14.
❒ May 6, 2024: XtalOpt Version 13.0 Released
- Added the multi-objective evolutionary search (MOES) functionality to search for novel functional materials through:
(1) optimization of a desired set of user-specified objectives (including the AFLOW-ML hardness),
(2) constrained evolutionary search. - Implemented the "localQueue" option to run XtalOpt locally on a computational cluster with local optimizations submitted to a queue.
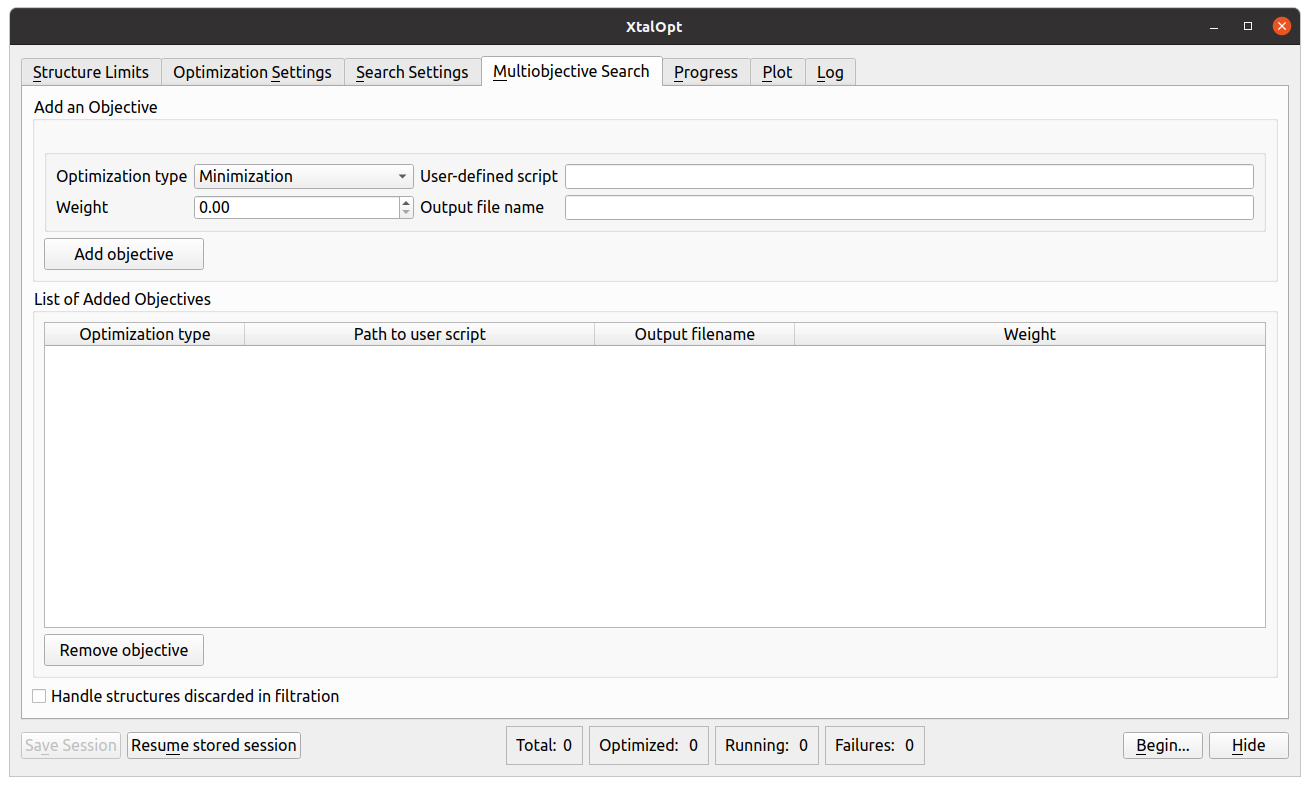
MOES tab in the graphical user interface of XtalOpt version 13.
- Added the "scaled volume" option to set the minimum and maximum volume limits for the search.
- Added the "softExit" and "hardExit" options to command-line interface for terminating the XtalOpt run once desired number of structures are generated.
- Added the support for the VASP machine learning OUTCAR output files.
- Various bug fixes.
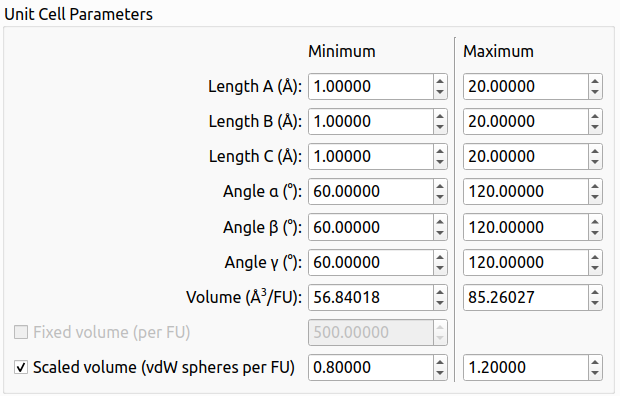
Setting the volume limits using the scaled volume option in the graphical user interface of XtalOpt version 13.
❒ Oct 21, 2018: XtalOpt Version r12.0 Released
- Added a hardness calculation via AFLOW-ML (Automatic FLOW for Materials Discovery - Machine Learning).
- Added a hardness fitness function, which allows for the prediction of hard structures.
- Added a generic optimizer, which allows the user to employ many previously unsupported optimizers for minimizing the geometry of an extended system.
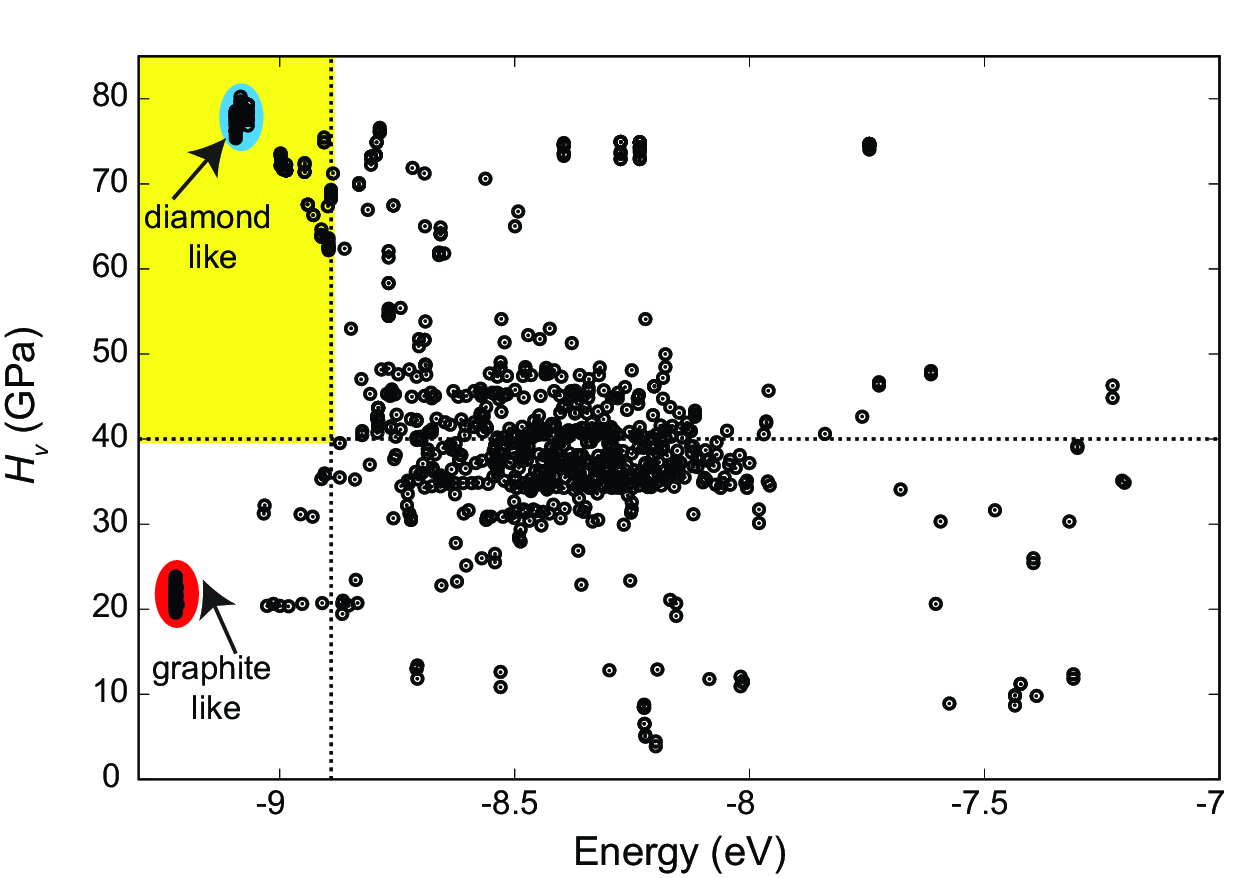
Example of a Hardness vs. Energy plot generated during a hardness search with XtalOpt.
- Added the ability to generate a simulated XRD (X-Ray Diffraction) pattern.
- Added the ability to use different optimizers and queuing interfaces for each optimization step.
- Implemented various bug fixes.
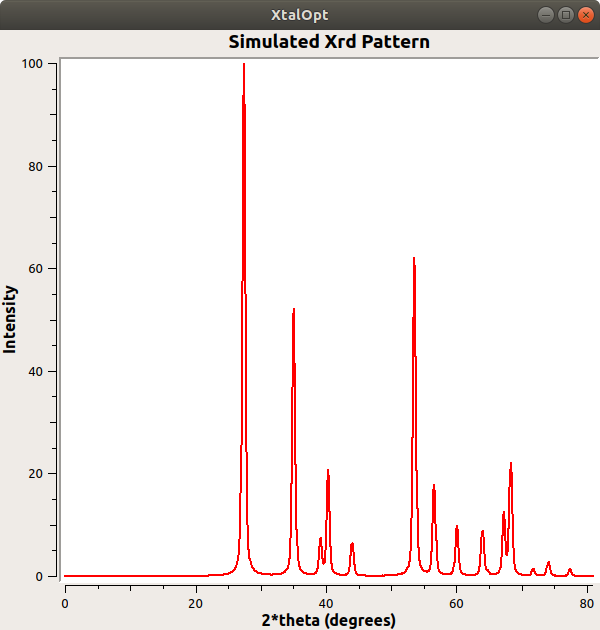
Example of a simulated XRD pattern generated with XtalOpt.
❒ Sep 11, 2017: XtalOpt Version r11.0 Released
- Removed dependence on Avogadro and Open Babel, making XtalOpt a stand-alone program rather than an extension.
- Changed the license from GPLv2 to a 3-Clause BSD license.
- Added the optional use of Avogadro2 to render crystals through a remote procedure call (RPC) protocol.
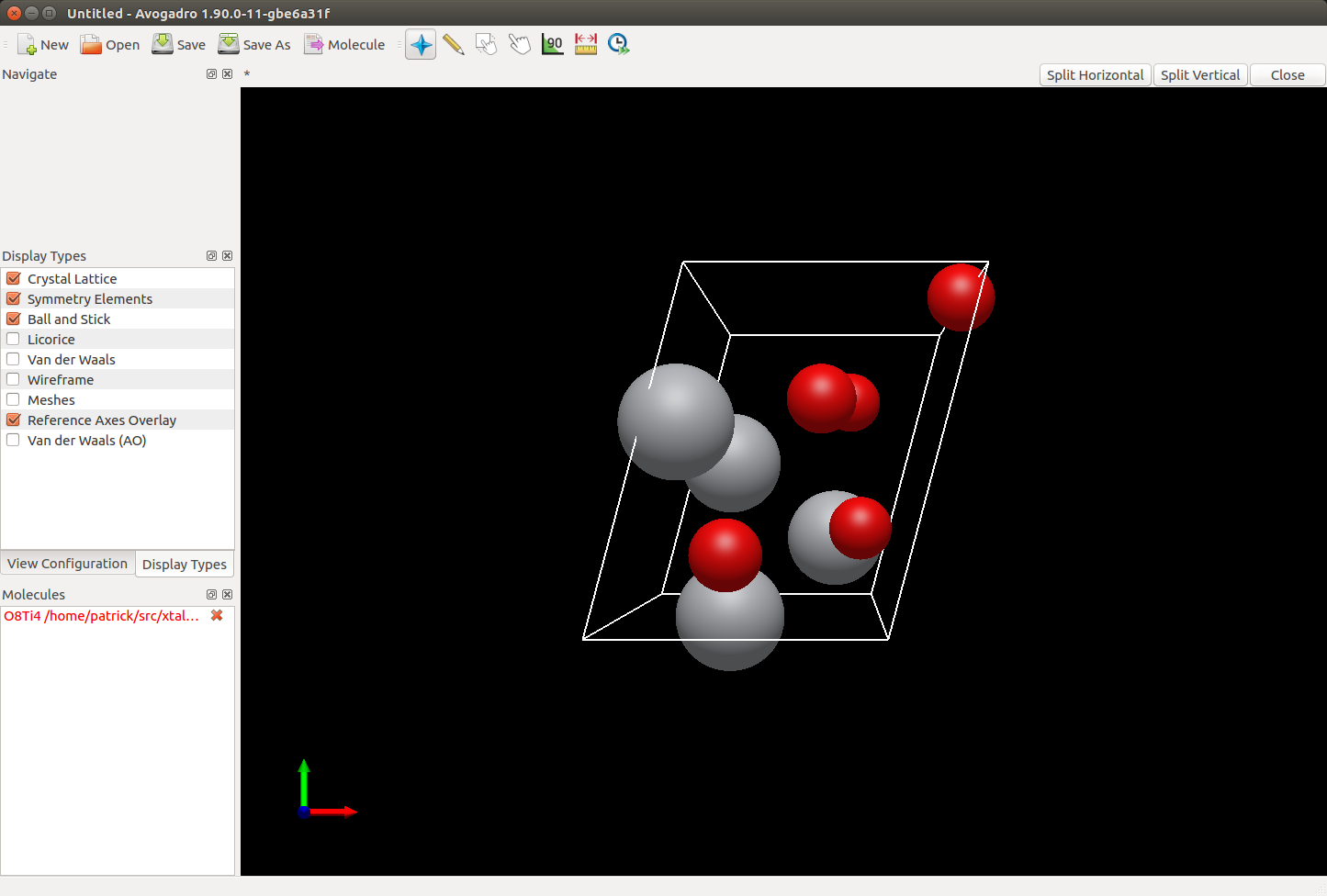
Example of using Avogadro2 to render a crystal via RPC.
- Added a command-line interface (CLI) to run the program and generate plots.
- Added the ability for the user to define custom minimum inter-atomic distances (IAD) between pairs of atom types.
- Implemented various bug fixes.
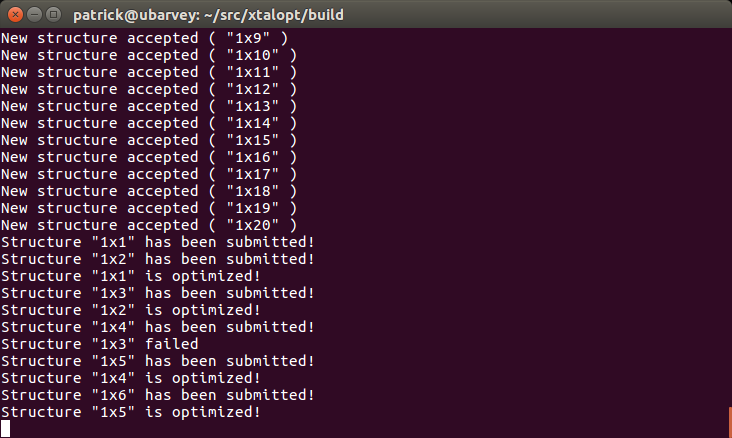
Example of using the CLI mode of XtalOpt.
❒ Jun 14, 2017: XtalOpt Version r10.0 Released
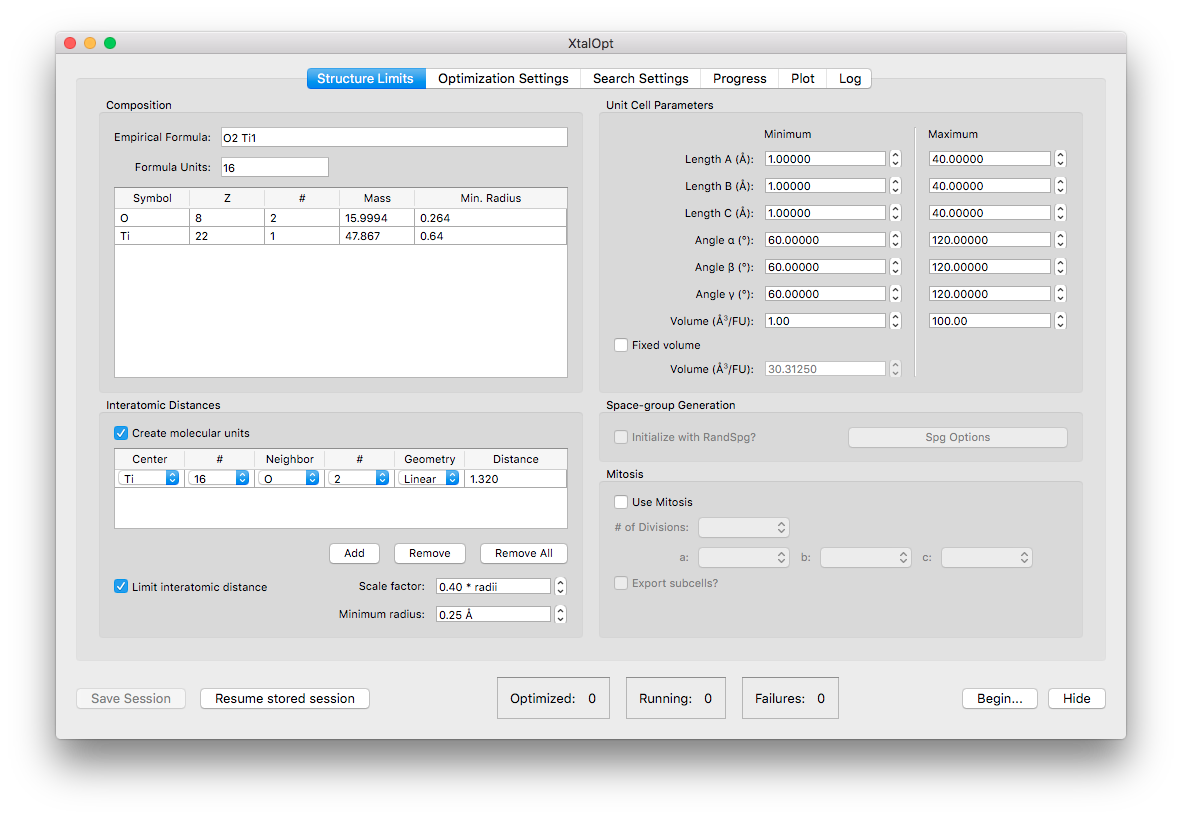
- Implementation of RandSpg, an algorithm that generates random crystals with specific spacegroups. This algorithm can optionally be used to create symmetric structures in the initial random generation.
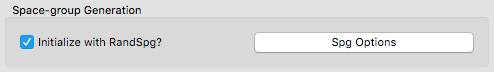
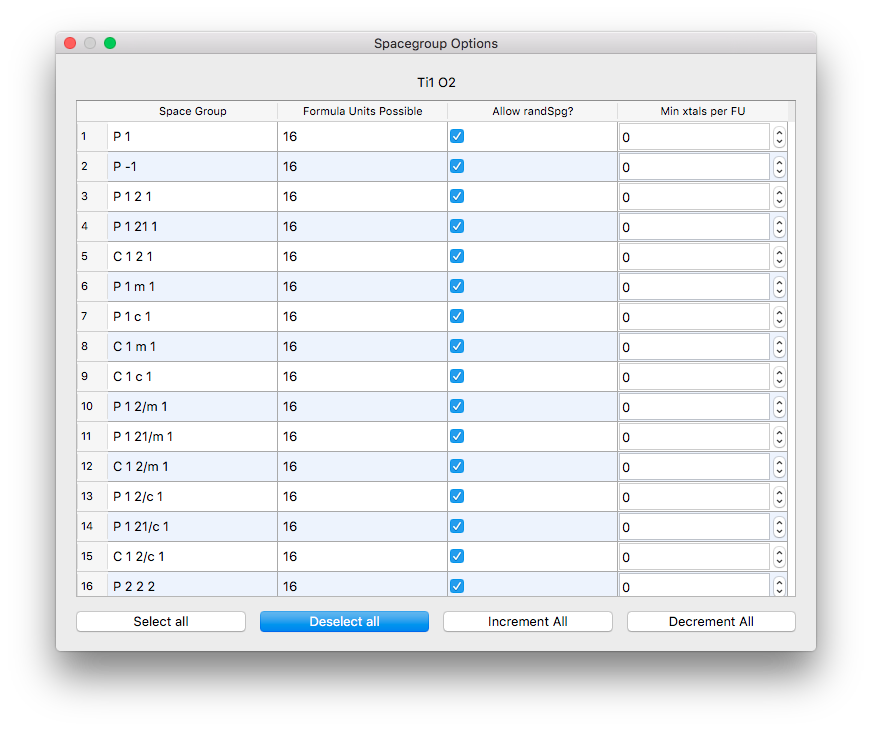
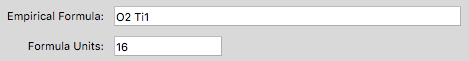
- Inclusion of variable formula units within XtalOpt enables the search for cells with multiple numbers of formula units within a single run.
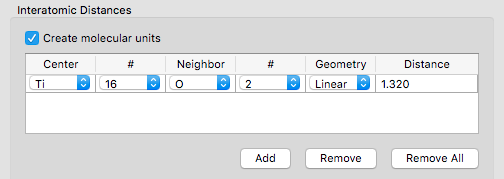
- A molecular-unit generator permits users to create single-center molecules in the unit cell during the initial generation step.
❒ Avogadro2 Updates
Our Ph.D. student, Patrick Avery, was accepted to the Google Summer of Code 2016!!!
He has been working on Avogadro2.
You can read about his most recent work on his blog.
❒ Aug 11, 2016: XtalOpt Version r9.1 Released
- Customizable polling interval for updating remote queue information.
- Automatic removal of remote working files optional.
- Optional removal of unnecessary files for VASP calculations.
- Added a new "mitosis" function used to generate higher local order for initial structures.
- Option to rank all current structures and export structures to a new subdirectory as .cml, CONTCAR, or .got.
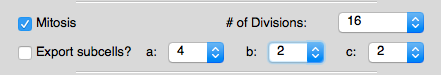
Options for the "mitosis" function found in the "Structure Limits" tab.
- Limit the total number of structures created during a run.
- New option to replace a failing structure with a new offspring.
- Ability to “inject” (seed) a structure mid-run
- Minimum atomic separation now specified as a fraction of the sum of atomic radii, with a hard minimum.
- Submission of remote calculations is throttled to ease DRMS load.
- More server-friendly method of fetching queue data.
- Support for GULP shell/core calculations added.
- Incorporated the XtalComp library for duplicate structure removal (niching).
- Fix compilation against Qt 4.6.3 and 4.8.0.
- Updated space-group detection library to spglib 1.0.8.
- Bundled libssh library removed, now an optional dependency.
- Added option to use command-line ssh/scp interfaces when libssh is unavailable or Kerberos authentication is needed.
- Implemented an extension for an automated stochastic docking program (RandomDock) that supports MOPAC, ADF, GAMESS and Gaussian as back-end molecular quantum chemistry engines.
- Numerous misc bug fixes.
Supporters
